[dropcap]H ypoprothrombinemia is a deficiency of prothrombin (clotting factor II) in the blood that is characterized by impaired hemostasis in response to trauma or a laceration.
Q: What is hemostasis and how is it altered by a deficiency of prothrombin?
A: Hemostasis encompasses the tightly regulated processes of blood clotting, platelet activation, and blood vessel repair.1
Prothrombin is a protein clotting factor present in blood that is involved in the first part of hemostasis, which is blood clotting or coagulation. Vitamin K is required for prothrombin production.
When a laceration or wound is sustained, prothrombin is converted to the enzyme thrombin. Thrombin in turn acts on fibrinogen to convert it to fibrin which then forms the framework of a clot to stop bleeding. Deficiency of prothrombin prevents this series of events and bleeding is not properly stopped.
After the clotting process of hemostasis would come the second part, platelet activation. Eventually, coagulation and platelet activation are switched off by blood-borne inhibitors.
What Is Hypoprothrombinemia In Celiac Disease and/or Gluten Sensitivity?
Sources:
Versteeg HH, Heemskerk JWM, Levi M, Reitsma PH. New Fundamentals in Hemostasis. Physiological Reviews Published 1 January 2013Vol. 93no. 327-358DOI: 10.1152/physrev.00016.2011 [↩]
Microscopic View of Hemochromatosis Stained Blue. Courtesy Wikimedia
What Is Hemochromatosis?
[dropcap]H emochromatosis, also called iron overload liver disease, is a common inherited disease in the Caucasian population that is characterized by increased iron deposition within the tissues (overload) associated with injury to them.
Hemochromatosis is an autosomal recessive disease, meaning a pair of abnormal genes are inherited from each parent. Parents are unaffected because they carry only one gene.
Q: Where is iron deposited?
A: In hemochromatosis, more iron is absorbed from the small intestine than is needed by the body. Subsequently, because the body has no satisfactory means to release iron overload, excess iron is deposited in various organs such as the liver causing cirrhosis, joints causing arthritis, and the pancreas causing diabetes mellitus.1
Consumption of alcoholic drinks with food should be avoided because alcohol increases leaky gut and greatly increases the absorption of iron. On the flip side, eggs and foods containing calcium such as milk and cheese are beneficial for hemochromatosis because they impair the absorption of iron. Another iron inhibitor is food that contains phytic acid which includes the bran and outer layer of grains, seeds, nuts, peas, beans, and lentils.
Iron overloading, as measured by a random (non-fasting) elevated transferrin saturation value, is estimated to occur in 1 to 6 people per 100 in the United States according to the CDC (Center for Disease Control and Prevention). Too much iron increases the risk for metabolic syndrome, type 2 diabetes mellitus, cancer, liver disease, and osteoporosis.
Medical treatment to remove excess iron from the body uses phlebotomy that draws blood through a vein and/or certain injectable drugs (deferoxamine and deferairox) that bind to iron.
What Is Hemochromatosis In Celiac Disease and/or Gluten Sensitivity?
[dropcap]O besity is an inflammatory metabolic disorder that is characterized by body mass index greater than 30% resulting from excessive body fat stored in adipose tissue.
Q: What is body fat?
A: Body fat is part of the body that functions as a reserve of stored energy. It is composed of fat cells, called adipocytes, having thin membranes between these cells. Adipocytes expand to store fat and shrink as fat is released as needed into the bloodstream for other body cells to use for metabolizing energy.
Each adipocyte contains a drop of triglyceride which is a type of lipid (fat). Triglycerides are a normal component in the bloodstream and, as such, are transported wherever needed as a form of energy. Excess triglycerides are the form of fat that is stored.
Initially, fat that is eaten in the diet is changed by digestive enzymes into the triglyceride form which is a molecule composed of three fatty acids and glycerol. Triglycerides are then absorbed through the small intestinal wall to be delivered to the liver. Of note, the liver can make triglycerides from excess protein and carbohydrates eaten in a meal, especially sugar and alcohol. The liver on the other hand makes cholesterol from triglycerides.
Triglyceride levels in the blood generally increase as weight increases. It is thought that an elevated blood triglyceride level hampers the body’s ability to feel full or satisfied with food that is eaten. Elevated triglyceride levels also increase the risk of clot formation because they cause the blood to become thicker. A normal triglyceride blood level is 150 mg/dL.
The causes of obesity are complex and varied. Those related to gluten sensitivity are discussed below.
What Is Obesity In Celiac Disease and/or Gluten Sensitivity?
Gluten sensitive enteropathy is active celiac disease characterized by inflammation of the small intestinal mucosa that results from an inherited immunologic intolerance to ingested gluten.
Q: What does the inflammation do to the mucosa in the small intestine?
A: Inflammation is a cell level immune response to gluten that has these effects on the mucosa:
Damages the barely visible villi (multitudinous finger-like structures) by causing atrophy or loss.
Likely affects the structural support and microcirculation of the villus, leading to collapse of the villus.
Elongates the crypts between villi. The thickening of the crypt is not so much a response to loss of surface enterocytes but represents inflammation of the mucosa.1
Increases round cells in the lamina propria and surface epithelial cells leaving few, irregular microvilli (brush border) on the surface of villi.
Damage is most intense in the duodenum and decreases toward the large intestine.
The extent of the damage to the intestine determines the malabsorptive consequences of the disease. Both gastric and small intestinal permeability are disrupted in patients with celiac disease.2
Relationship between active celiac disease and intestinal permeability: There is a clear association between degree of mucosal damage and the intestinal-permeability ratio, and a normal ratio generally implies near-normal small intestinal structure. A raised intestinal permeability of the mucosal lining (leaky gut) could predispose to a high absorption of gluten and exacerbate an existing lesion and hence convert a latent to an overt enteropathy.3
Relationship between active celiac disease and tight junction proteins: A study of intestinal permeability showed that the expression of all junction proteins of the small intestinal lining (occludin, claudin 3, zonula occludens 1, and E-cadherin) was already decreased in early stage celiac disease when compared with non-celiac controls, showing leaky gut and confirming the above earlier study by Johnston et al. Junction protein expression correlated positively with mucosal villus structure and negatively with the number of intraepithelial lymphocytes (IELs), the intensity of small-intestinal autoantibody deposits, and serum autoantibodies. The expression of claudin 3 showed a negative correlation with diarrheal score.4
Relationship between active celiac disease and inflammation. In celiac disease there is an over production of inflammatory interleukin-15 (IL-15) which inhibits the correct removal of damaged intraepithelial lymphocytes caused by the reaction to gluten. Serum levels of IL-15 are directly correlated with the seriousness of tissue damage.5
Relationship between active celiac disease and gut microbiota. Results of a study investigating intestinal microbiota (normal bacterial residents) in patients with celiac disease suggest that with lower levels of the genus bifidobacteria, celiac patients have an imbalance in the intestinal microbiota even while on a gluten-free diet. This fact could favor the pathological process of the disorder. The concentration of bifidobacteria per gram of feces was significantly higher in healthy subjects (2.5 ± 1.5 x107 CFU/g) when compared to celiac patients (1.5 ± 0.63 x108 CFU/g).6
Relationship between active celiac disease and endoscopy technique. The most severe degree of villous atrophy was detected when distal duodenal biopsy specimens were taken in addition to a duodenal bulb biopsy specimen from either the 9- or 12-o’clock position (96.4% sensitivity; 95% CI, 79.7%-100%). The difference between the 12-o’clock position biopsy and the 3-o’clock position biopsy in detecting the most severe villous atrophy was 92% (24/26 patients) versus 65% (17/26 patients).7
Relationship between active celiac disease and diet adherence. Patients with consistent gluten free diet adherence experience symptomatic responses to dietary gluten (SRDG) faster and more severe in comparison to their prior gluten exposure possibly demonstrating an adept immunological response. Anxiety and depression also enhance the speed of symptom onset and co-existing visceral hypersensitivity is a risk factor for severe reactions to dietary gluten.8
Relationship between active celiac disease and atrial fibrillation: Patients with celiac disease, verified by intestinal biopsy, are at increased risk of atrial fibrillation. This observation is consistent with previous findings that elevation of inflammatory markers predicts atrial fibrillation.9
How Prevalent Is Gluten Sensitive Enteropathy?
Sources:
Murray JA, the widening spectrum of celiac disease. American Journal of Clinical Nutrition. Mar 1999; 69(3):354-365. [↩]
Murray JA, the widening spectrum of celiac disease. American Journal of Clinical Nutrition. Mar 1999; 69(3):354-365. [↩]
Johnston SD, Smye M, Watson RGP. Intestinal permeability and morphometric recovery in coeliac disease. Lancet. Jul 28, 2001;358(9278):259, 2p. [↩]
Rauhavirta T, Lindfors K, Koskinen O, Laurila K, Kurppa K, Saavalainen P, Mäki M, Collin P, Kaukinen K. Impaired epithelial integrity in the duodenal mucosa in early stages of celiac disease. Transl Res. 2014 Sep;164(3):223-31. doi: 10.1016/j.trsl.2014.02.006 [↩]
Stazi AV, Trinti B. Selenium status and over-expression of interleukin-15 in celiac disease and autoimmune thyroid diseases. Ann Ist Super Sanita. 2010;46(4):389-99.DOI: 10.4415/ANN_10_04_06. [↩]
Golfetto L, de Senna FD, Hermes J, Beserra BT, França Fda S, Martinello F. Lower bifidobacteria counts in adult patients with celiac disease on a gluten-free diet. Arq Gastroenterol. 2014 Apr-Jun;51(2):139-43. [↩]
Kurien M, Evans KE, Hopper AD, Hale MF, Cross SS, Sanders DS. Duodenal bulb biopsies for diagnosing adult celiac disease: is there an optimal biopsy site? Gastrointest Endosc. 2012 Jun;75(6):1190-6. doi: 10.1016/j.gie.2012.02.025. [↩]
Barratt SM, Leeds JS, Sanders DS. Factors influencing the type, timing and severity of symptomatic responses to dietary gluten in patients with biopsy-proven coeliac disease. J Gastrointestin Liver Dis. 2013 Dec;22(4):391-6. [↩]
Emilsson L, Smith JG, West J, Melander O, Ludvigsson JF. Increased risk of atrial fibrillation in patients with coeliac disease: a nationwide cohort study. Eur Heart J. 2011 Oct;32(19):2430-7. doi: 10.1093/eurheartj/ehr167. [↩]
[dropcap]L ymphadenopathy is an alteration of lymph nodes that is characterized by enlargement of lymph nodes greater than 1.5 cm caused by proliferation (increased production) of lymphocytes within the node.
Q: What are lymph nodes?
A: Lymph nodes are part of the lymphatic system, acting to protect body fluids by filtering out and destroying bacteria and other harmful substances from lymph that is continually carried to them by lymph vessels.
Cleaned lymph is carried away from the nodes by lymph vessels to the bloodsteam where it helps forms blood plasma. Lymph nodes produce various blood cells needed to fight infection which includes lymphocytes.
Lymphocytes are small white blood cells that plays a major role in defending the body against disease. There are two types of lynphocytes: B cells, which make antibodies that attack bacteria and toxins, and T cells which attack body cells themselves when they have been taken over by viruses or become cancerous.
Lymph nodes that become enlarged doing battle with an infection or as a result of injury nearby usually resolve with treatment of the infection or injury. However, if the cause is cancer, the nodes would need to be treated as well as the cancer.
What Is Lymphadenopathy In Celiac Disease and/or Gluten Sensitivity?
Parathyroid Glands in the Neck. Courtesy Wikipedia.com
What Is Secondary Hyperparathyroidism?
[dropcap]S econdary hyperparathyroidism is a parathyroid disorder resulting from hypocalcemia (low blood calcium level) that is characterized by excessive production of parathyroid hormone in the attempt to normalize the low blood calcium by releasing calcium from bone.
Parathyroid hormone is produced by the four pea sized parathyroid glands that are located on the thyroid gland in the front of the neck. In part, because the thyroid and parathyroid glands share the same anatomic place in the body and because they have similar names, they are often confused although they have completely different actions.
Parathyroid hormone regulates calcium and the opposing mineral phosphorus in the blood. In secondary hyperparathyroidism, calcium blood levels are low to normal while phosphorus levels are increased which stimulates the outpouring of parathyroid hormone.
Q: How does secondary hyperparathyroidism differ from primary hyperparathyroidism?
A: In primary hyperparathyroidism blood calcium is high and phosphorus is low, which is the opposite of secondary hyperparathyroidism.
The most common cause of secondary hyperparathyroidism is kidney disease causing failure to reabsorb calcium followed by vitamin D deficiency and malabsorption.
What Is Secondary Hyperparathyroidism In Celiac Disease and/or Gluten Sensitivity?
[dropcap]A[/dropcap]nti-endomysium antibodies (EmA) are connective tissue autoantibodies produced in persons who have inherited the genes for celiac disease, an autoimmune disease, and are reacting to gluten in their diet.
Autoantibodies are abnormal in that they attack the body’s own tissue.
Q: What is endomysium?
A: Endomysium is the delicate connective tissue that surrounds individual muscle fibers. The autoantigen, or target, that stimulates the autoimmune response is the naturally occuring enzyme in endomysium called tissue transglutaminase (tTG), or more specifically tranglutaminase-2 (TG2). Anti-tissue transglutaminase antibodies recognize the same antigen as EmA, from which they differ in terms of detection method.
Anti-endomysium antibodies (EmA) are tested by the indirect immunofluorescence method and directed against “reticulin-like” fibres in connective tissue around smooth muscle fibres in the esophagus, liver, stomach, and bladder of monkeys, in the sections of the jejunum and kidneys of rats and in sections of the human umbilical cord. In comparison, for the determination of anti-tissue transglutaminase IgA and IgG antibodies, ELISA with human extractive or recombinant transglutaminase is recommended.1
EmA‐binding patterns in serum samples from patients with celiac disease have proved to be exclusively TG2‐targeted and the correlation between EmA and TG2 antibodies is therefore good. Evidence shows that celiac autoantibodies are produced in the small‐bowel mucosa.2
What Are Anti-Endomysium Antibodies In Celiac Disease and/or Gluten Sensitivity?
Sources:
Trigoni E, Tsirogianni A, Pipi E, Mantzaris G, Papasteriades C. Celiac disease in adult patients: specific autoantibodies in the diagnosis, monitoring, and screening. Autoimmune Dis. 2014;2014:623514. doi: 10.1155/2014/623514. [↩]
Salmi TT, Collin P, Korponay-Szabó IR, Laurila K, Partanen J, Huhtala H, Király R, Lorand L, Reunala T, Mäki M, Kaukinen K. Endomysial antibody‐negative coeliac disease: clinical characteristics and intestinal autoantibody deposits.Gut. 2006 Dec;55(12):1746-53. [↩]
[dropcap]M uscle spasm and muscle cramps are disorders of muscle function caused by painful involuntary contractions of affected skeletal muscles characterized by limited movement.
Q: What is the difference between muscle spasm and cramps?
A: Cramps are stronger and more painful than spasms, occurring while the muscle is in its most shortened state.
Skeletal muscles are those that move the body skeleton to do work as we choose. Examples are muscles that move the mouth, arms, hands, legs, and feet.
What Are Muscle Spasm, Muscle Cramps In Celiac Disease and/or Gluten Sensitivity?
[dropcap]M uscle wasting, or atrophy, is a muscle disorder resulting from the loss of muscle tissue which is characterized by thin muscles that have reduced strength and endurance.
What Is Muscle Wasting In Celiac Disease and/or Gluten Sensitivity?
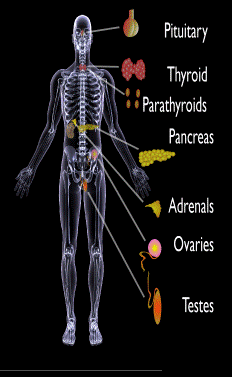
Endocrine glands targeted in polyglandular autoimmune syndrome.
What Are Autoimmune Polyglandular Syndromes?
[dropcap]A utoimmune polyglandular syndromes (APS) are rare clusterings of two or more endocrine and non-endocrine autoimmune disorders in the same affected person.
Polyglandular is somewhat of a misnomer since many of the manifestations of the diseases do not concern endocrine glands.1
Endocrine autoimmune disorders involve the abnormal production of autoantibodies that target and destroy the body’s own endocrine tissues, causing loss of essential hormone production by the targeted glands. Endocrine glands include the pituitary, thyroid, adrenal, parathyroid, islets of Langerhans (pancreas), testes in males, and ovaries in females.
First degree relatives (siblings of same parents, parents, children) have an increased incidence of latent, meaning not apparent, autoimmune pathology.2
Q: How many autoimmune polyglandular syndromes are described?
A:Three syndromes have been identified and they are all inherited: APS type-1, APS type-2, and APS type-3.
APS type-1 is a genetic mutation inherited in an autosomal recessive manner. A child with APS type-1 has inherited two mutated copies of a gene called the AIRE (autoimmune regulator) gene, which is on the long arm of 21st chromosome present in each cell.3 The parents, called carriers, are unaffected since they each have only a single copy of the AIRE mutated gene. Humans have a total of 23 pairs of chromosomes that contain genes inherited from each parent. Mutations in the genes cause disease.
Diagnosis criteria for autoimmune polyglandularsyndrome type-1 includes these three disorders:
Chronic candida infection (CMC), which usually develops first, typically attacks skin, but very commonly also nails, mouth, vagina, esophagus and intestine. CMC in APS type-1 patients is usually mild, and in most cases, it is chronic. It is found in 73–100 % of APS type-1 patients.
Hypoparathyroidism, causing loss of parathyroid function (hypoparathyreosis) is found in 76–93 % of APS type-1 patients.
Autommune Addison’s disease, also called autoimmune adrenalitis, is found in 72-100 % of APS type-1 patients. Still many of them die for unrecognized or late diagnosed autoimmune Addison’s disease, so regular follow-up for children in suspicion of APS type-1 (with CMC or/and hypoparathyroidism) is necessary.4
Other assocated disorders that may develop, but are not required for diagnosis, include: vitiligo, premature menopause, pernicious anemia, parathyroid gland failure, alopecia, and celiac disease. Thyroid disease rarely occurs.5
APS type-2 is linked to the inheritance of HLA antigens on chromosome 6 and appears to be autosomal dominant with incomplete penetrance. This suggests the contribution of environmental factors, such as bacterial and viral infections, medications, psychological factors, etc.6It does not have an identified mutation of the AIRE gene.
Diagnosis criteria for autoimmune polyglandular syndrome type-2 includes these two disorders:
Autommune Addison’s disease combined with
Autoimmune thyroid disease (thyroid atrophy, hypertrophic goiter related to Hashimoto’s thyroiditis, Graves’ disease, asymptomatic autoimmune thyroiditis)6 and/or type I diabetes mellitus. The conditions may occur in any order.
Polyglandular autoimmune syndrome type-2 is also known as Schmidt’s syndrome when adrenalitis (adrenal insufficiency) is associated with thyroiditis and Carpenter’s syndrome for adrenal insufficiency with hypoparathyreosis (impaired function of parathyroid glands).
Other disorders that may develop, but are not required for diagnosis, include: type 1 diabetes (50%), frequently gonadal failure or vitiligos, also celiac disease, autoimmune hepatitis, alopecia, pernicious anemia, and myasthenia gravis.7 Decades may arise between the onset of one disease and the onset of the second in the same patient.8
Therapy of APS type-2 consists of hormone replacement therapy for each separate condition, except that treatment for adrenal insufficiency must be given before thyroid therapy is started when the conditions occur together.9 Thyroxin replacement may induce life-threatening adrenal failure in a patient with untreated Addison’s disease. Thus, in case of doubt hydrocortisone should be given before the thyroxine administration is started.10
APS type-3 has a strong genetic background. Diagnosis criteria for autoimmune polyglandular syndrome type-3 involves these conditions:
Autoimmune thyroiditis that occurs with another organ-specific autoimmune disease, but not with autoimmune Addison’s disease, and
Other autoimmune diseases can include diabetes mellitus, pernicious anemia, vitiligo, alopecia, myasthenia gravis, celiac disease, and Sjögren’s syndrome. The most common APS type-3 combination is autoimmune disease of thyroid gland and pernicious anemia.11
Who is Affected in the General Population?
APS type-1 is usually apparent in childhood with the incidence of 1 in100,000 persons. It is more common among Finns (1 in 25,000), Sardinians (1 in 14,000), and Iranian Jews (1 in 6,500 to 1 in 9,000). The age of onset is usually early childhood, but new symptoms can develop throughout life. It affects both sexes equally.
APS type-2 has a peak onset in middle age, although the first signs usually develop between 20–30 years of age. Its prevalence is 1 in 20,000 persons. It is three times more frequent among women than men.12
APS type-3 is most frequent among middle-aged women.7[/box]
What Is Autoimmune Polyglandular Syndrome In Celiac Disease and/or Gluten Sensitivity?
Sources:
Wémeau JL, Proust-Lemoine E, Ryndak A, Vanhove L. Thyroid autoimmunity and polyglandular endocrine syndromes. Hormones (Athens). 2013 Jan-Mar;12(1):39-45. [↩]
Femiano P, Castaldo V, Iossa C. Complex family association in autoimmune polyendocrine syndrome. Minerva Pediatrica. Apr 2003;55(2):163-70. [↩]
Wémeau JL, Proust-Lemoine E, Ryndak A, Vanhove L. Thyroid autoimmunity and polyglandular endocrine syndromes. Hormones (Athens). 2013 Jan-Mar;12(1):39-45. [↩]
Wémeau JL, Proust-Lemoine E, Ryndak A, Vanhove L. Thyroid autoimmunity and polyglandular endocrine syndromes. Hormones (Athens). 2013 Jan-Mar;12(1):39-45. [↩]
Wémeau JL, Proust-Lemoine E, Ryndak A, Vanhove L. Thyroid autoimmunity and polyglandular endocrine syndromes. Hormones (Athens). 2013 Jan-Mar;12(1):39-45. [↩] [↩]
MAJERONI BA and PATEL P. Autoimmune Polyglandular Syndrome, Type II. Am Fam Physician. 2007 Mar 1;75(5):667-670. [↩]
Lipowsky C, Schorl-Schweikardt BA, Kehl O, Brändle M. 19-year-old patient with adrenal cortex insufficiency–only the tip of the iceberg. Polyendocrine autoimmune syndrome type II (Schmidt syndrome). Praxis (Bern 1994). 2008 Jan 23;97(2):77-81. [↩]
Van den Driessche A, Eenkhoorn V, Van Gaal L, De Block C. Type 1 diabetes and autoimmune polyglandular syndrome: a clinical review. Neth J Med. 2009 Dec;67(11):376-87. [↩]
 What Is Hypoprothrombinemia?
What Is Hypoprothrombinemia?