Underarm showing skin darkening, which is a feature of Addison’s Disease
What Is Primary Addison’s Disease?
[dropcap]A[/dropcap]ddison’s disease is an autoimmune destruction of the adrenal glands by autoantibodies that target the adrenal cortex, or outer part of these glands, and is characterized by a slow progressive failure of the adrenal glands to adequately produce its steroid hormones.
Symptoms of adrenal fatigue or failure may not develop until the majority of adrenal tissue is destroyed. When untreated, progression leads to coma, called Addisonian crisis, which is a medical emergency.
There are two adrenal glands each located on top of a kidney and enclosed in a connective tissue capsule. Each is a small, triangular shape that is made of two parts: the outer region and the inner region.
The inner region, called the adrenal medulla, produces epinephrine and norepinephrine chemicals that are needed to deal with stress.
The outer region, called the adrenal cortex, produces adrenocortical (steroid) hormones and releases them into the bloodstream in response to pituitary stimulating hormone from the brain.
Q: What is the function of steroid hormones produced by the adrenal glands?
A: Functions of the three steroid hormones produced by the adrenal glands are:
Glucocorticoids restrain inflammation and metabolism of carbohydrates, fats and proteins to maintain a normal glucose blood level. The major glucocorticoid is hydrocortisone.
Mineralocorticoids regulate the retention and excretion of fluids and electrolytes by the kidneys. The most important mineralocorticoid is aldosterone.
Androgen (testosterone) is a male sex hormone.
Secondary adrenal insufficiency may develop from other causes that are not immune related such as chronic infections, tumor, and medications.
What Is Addison’s Disease In Celiac Disease and/or Gluten Sensitivity?
What Is Vitamin B12 Deficiency Anemia? [dropcap]V itamin B12 deficiency anemia is a megaloblastic anemia that is characterized by defective DNA synthesis of red blood cells due to a lack of vitamin B12. Vitamin B12 is…
[dropcap]L ymphadenopathy is an alteration of lymph nodes that is characterized by enlargement of lymph nodes greater than 1.5 cm caused by proliferation (increased production) of lymphocytes within the node.
Q: What are lymph nodes?
A: Lymph nodes are part of the lymphatic system, acting to protect body fluids by filtering out and destroying bacteria and other harmful substances from lymph that is continually carried to them by lymph vessels.
Cleaned lymph is carried away from the nodes by lymph vessels to the bloodsteam where it helps forms blood plasma. Lymph nodes produce various blood cells needed to fight infection which includes lymphocytes.
Lymphocytes are small white blood cells that plays a major role in defending the body against disease. There are two types of lynphocytes: B cells, which make antibodies that attack bacteria and toxins, and T cells which attack body cells themselves when they have been taken over by viruses or become cancerous.
Lymph nodes that become enlarged doing battle with an infection or as a result of injury nearby usually resolve with treatment of the infection or injury. However, if the cause is cancer, the nodes would need to be treated as well as the cancer.
What Is Lymphadenopathy In Celiac Disease and/or Gluten Sensitivity?
Microscopic Image Showing a Pink Collagen Band in Collagenous Colitis.
What Is Collagenous Colitis?
[dropcap]C ollagenous colitis is a disease of the large intestine (colon) that is characterized by microscopic inflammation of the surface mucosal lining and an abnormally thickened collagen band of tissue that develops wthin the lining of the colon.
The thicker than normal layer of collagen of at least 10 µm (reference value: 2–7 µm) can vary in different locations. Inflammation occurs with increased numbers of lymphocytes (white blood cells) and plasma cells and epithelial (surface cell) damage. These changes can only be seen under microscopic examination of multiple biopsied tissue samples taken during a colonoscopy procedure.
Q: What is collagen?
A:Collagen is a strong, fibrous protein found in connective tissue of the colon and many other tissues such as tendons. The normal basement membrane in the bowel consists mainly of collagen type IV, laminin, and fibronectin. The increased collagen band observed in collagenous colitis consists basically of collagen type I and III, which are the subtypes produced by repair functions, indicating a reactive origin to some irritant or drug.1
The biopsies should preferably be taken from the ascending colon, since the pathological hallmarks may be absent in the descending colon, and in the normally occurring thicker collagen layer in the rectosigmoid region.1 Inflammation of the ileum (last segment of the small intestine next to colon) is common.2
Endoscopy and radiological (x-ray) examinations are usually normal.3
Autoimmune disorders are frequently seen in adult patients with collagenous colitis.4 In the study below by Koskela et al. concomittent autoimmune diseases were present in 53% of patients with collagenous colitis.5
Importantly, the finding of collagenous colitis in patients with autoimmune diseases may reflect the treatment with NSAIDs (non-steroidal anti-inflammatory drugs), such as Ibuprofin and aspirin, PPIs (proton pump inhibitors), and other drugs. However, if secondary forms of collagenous colitis are not taken into consideration, underlying, treatable diseases may be overlooked, while only the gastrointestinal symptoms are treated symptomatically or with budesonide (a steroid).6
Treatment with budesonide steroid is efficacious irrespective of bile acid malabsorption.7
Budesonide at a mean dose of 4.5 mg/day maintained clinical remission for at least 1 year in the majority of patients with collagenous colitis and preserved health-related quality of life without safety concerns. Treatment extension with low-dose budesonide beyond 1 year may be beneficial given the high relapse rate after budesonide discontinuation.8
See below for nutritional deficiency problems caused by steroid usage and steps to be taken for correction.
What Is Collagenous Colitis In Celiac Disease and/or Gluten Sensitivity?
Sources:
Ohlsson B. New insights and challenges in microscopic colitis. Therap Adv Gastroenterol. 2015 Jan;8(1):37-47. doi: 10.1177/1756283X14550134. [↩] [↩]
Bjørnbak C, Engel PJ, Nielsen PL, Munck LK. Microscopic colitis: clinical findings, topography and persistence of histopathological subgroups. Aliment Pharmacol Ther. 2011 Nov;34(10):1225-34. doi: 10.1111/j.1365-2036.2011.04865.x. [↩]
Abdo AA, Urbanski SJ, Beck PL. Lymphotcytic and collagenous colitis: the emerging entity of microscopic colitis. An update on pathophysiology, diagnosis and management. Canadian Journal of Gastroenterology. Jul 2003;17(7):425-32. [↩]
Leung ST, Chandan VS, Murray JA, Wu TT. Collagenous gastritis: histopathologic features and association with other gastrointestinal diseases. Am J Surg Pathol. 2009 May;33(5):788-98. doi: 10.1097/PAS.0b013e318196a67f. [↩]
Koskela RM, Niemela SE, Karttunen TJ, Lehtola JK. Clinical characteristics of collagenous and lymphocytic colitis. Scandanavian Journal of Gastroenterology. Sep 2004;39(9):837-45. [↩]
Ohlsson B. New insights and challenges in microscopic colitis. Therap Adv Gastroenterol. 2015 Jan;8(1):37-47. doi: 10.1177/1756283X14550134. [↩]
Bjørnbak C, Engel PJ, Nielsen PL, Munck LK. Microscopic colitis: clinical findings, topography and persistence of histopathological subgroups. Aliment Pharmacol Ther. 2011 Nov;34(10):1225-34. doi: 10.1111/j.1365-2036.2011.04865.x. [↩]
Münch A, Bohr J, Miehlke S, et al. Low-dose budesonide for maintenance of clinical remission in collagenous colitis: a randomised, placebo-controlled, 12-month trial. Gut. 2014 Nov 25. pii: gutjnl-2014-308363. doi: 10.1136/gutjnl-2014-308363. [↩]
[dropcap]M uscle wasting, or atrophy, is a muscle disorder resulting from the loss of muscle tissue which is characterized by thin muscles that have reduced strength and endurance.
What Is Muscle Wasting In Celiac Disease and/or Gluten Sensitivity?
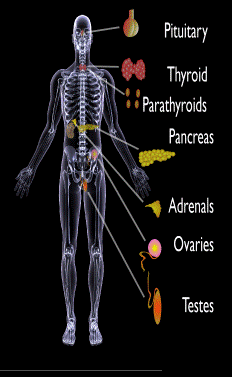
Endocrine glands targeted in polyglandular autoimmune syndrome.
What Are Autoimmune Polyglandular Syndromes?
[dropcap]A utoimmune polyglandular syndromes (APS) are rare clusterings of two or more endocrine and non-endocrine autoimmune disorders in the same affected person.
Polyglandular is somewhat of a misnomer since many of the manifestations of the diseases do not concern endocrine glands.1
Endocrine autoimmune disorders involve the abnormal production of autoantibodies that target and destroy the body’s own endocrine tissues, causing loss of essential hormone production by the targeted glands. Endocrine glands include the pituitary, thyroid, adrenal, parathyroid, islets of Langerhans (pancreas), testes in males, and ovaries in females.
First degree relatives (siblings of same parents, parents, children) have an increased incidence of latent, meaning not apparent, autoimmune pathology.2
Q: How many autoimmune polyglandular syndromes are described?
A:Three syndromes have been identified and they are all inherited: APS type-1, APS type-2, and APS type-3.
APS type-1 is a genetic mutation inherited in an autosomal recessive manner. A child with APS type-1 has inherited two mutated copies of a gene called the AIRE (autoimmune regulator) gene, which is on the long arm of 21st chromosome present in each cell.3 The parents, called carriers, are unaffected since they each have only a single copy of the AIRE mutated gene. Humans have a total of 23 pairs of chromosomes that contain genes inherited from each parent. Mutations in the genes cause disease.
Diagnosis criteria for autoimmune polyglandularsyndrome type-1 includes these three disorders:
Chronic candida infection (CMC), which usually develops first, typically attacks skin, but very commonly also nails, mouth, vagina, esophagus and intestine. CMC in APS type-1 patients is usually mild, and in most cases, it is chronic. It is found in 73–100 % of APS type-1 patients.
Hypoparathyroidism, causing loss of parathyroid function (hypoparathyreosis) is found in 76–93 % of APS type-1 patients.
Autommune Addison’s disease, also called autoimmune adrenalitis, is found in 72-100 % of APS type-1 patients. Still many of them die for unrecognized or late diagnosed autoimmune Addison’s disease, so regular follow-up for children in suspicion of APS type-1 (with CMC or/and hypoparathyroidism) is necessary.4
Other assocated disorders that may develop, but are not required for diagnosis, include: vitiligo, premature menopause, pernicious anemia, parathyroid gland failure, alopecia, and celiac disease. Thyroid disease rarely occurs.5
APS type-2 is linked to the inheritance of HLA antigens on chromosome 6 and appears to be autosomal dominant with incomplete penetrance. This suggests the contribution of environmental factors, such as bacterial and viral infections, medications, psychological factors, etc.6It does not have an identified mutation of the AIRE gene.
Diagnosis criteria for autoimmune polyglandular syndrome type-2 includes these two disorders:
Autommune Addison’s disease combined with
Autoimmune thyroid disease (thyroid atrophy, hypertrophic goiter related to Hashimoto’s thyroiditis, Graves’ disease, asymptomatic autoimmune thyroiditis)6 and/or type I diabetes mellitus. The conditions may occur in any order.
Polyglandular autoimmune syndrome type-2 is also known as Schmidt’s syndrome when adrenalitis (adrenal insufficiency) is associated with thyroiditis and Carpenter’s syndrome for adrenal insufficiency with hypoparathyreosis (impaired function of parathyroid glands).
Other disorders that may develop, but are not required for diagnosis, include: type 1 diabetes (50%), frequently gonadal failure or vitiligos, also celiac disease, autoimmune hepatitis, alopecia, pernicious anemia, and myasthenia gravis.7 Decades may arise between the onset of one disease and the onset of the second in the same patient.8
Therapy of APS type-2 consists of hormone replacement therapy for each separate condition, except that treatment for adrenal insufficiency must be given before thyroid therapy is started when the conditions occur together.9 Thyroxin replacement may induce life-threatening adrenal failure in a patient with untreated Addison’s disease. Thus, in case of doubt hydrocortisone should be given before the thyroxine administration is started.10
APS type-3 has a strong genetic background. Diagnosis criteria for autoimmune polyglandular syndrome type-3 involves these conditions:
Autoimmune thyroiditis that occurs with another organ-specific autoimmune disease, but not with autoimmune Addison’s disease, and
Other autoimmune diseases can include diabetes mellitus, pernicious anemia, vitiligo, alopecia, myasthenia gravis, celiac disease, and Sjögren’s syndrome. The most common APS type-3 combination is autoimmune disease of thyroid gland and pernicious anemia.11
Who is Affected in the General Population?
APS type-1 is usually apparent in childhood with the incidence of 1 in100,000 persons. It is more common among Finns (1 in 25,000), Sardinians (1 in 14,000), and Iranian Jews (1 in 6,500 to 1 in 9,000). The age of onset is usually early childhood, but new symptoms can develop throughout life. It affects both sexes equally.
APS type-2 has a peak onset in middle age, although the first signs usually develop between 20–30 years of age. Its prevalence is 1 in 20,000 persons. It is three times more frequent among women than men.12
APS type-3 is most frequent among middle-aged women.7[/box]
What Is Autoimmune Polyglandular Syndrome In Celiac Disease and/or Gluten Sensitivity?
Sources:
Wémeau JL, Proust-Lemoine E, Ryndak A, Vanhove L. Thyroid autoimmunity and polyglandular endocrine syndromes. Hormones (Athens). 2013 Jan-Mar;12(1):39-45. [↩]
Femiano P, Castaldo V, Iossa C. Complex family association in autoimmune polyendocrine syndrome. Minerva Pediatrica. Apr 2003;55(2):163-70. [↩]
Wémeau JL, Proust-Lemoine E, Ryndak A, Vanhove L. Thyroid autoimmunity and polyglandular endocrine syndromes. Hormones (Athens). 2013 Jan-Mar;12(1):39-45. [↩]
Wémeau JL, Proust-Lemoine E, Ryndak A, Vanhove L. Thyroid autoimmunity and polyglandular endocrine syndromes. Hormones (Athens). 2013 Jan-Mar;12(1):39-45. [↩]
Wémeau JL, Proust-Lemoine E, Ryndak A, Vanhove L. Thyroid autoimmunity and polyglandular endocrine syndromes. Hormones (Athens). 2013 Jan-Mar;12(1):39-45. [↩] [↩]
MAJERONI BA and PATEL P. Autoimmune Polyglandular Syndrome, Type II. Am Fam Physician. 2007 Mar 1;75(5):667-670. [↩]
Lipowsky C, Schorl-Schweikardt BA, Kehl O, Brändle M. 19-year-old patient with adrenal cortex insufficiency–only the tip of the iceberg. Polyendocrine autoimmune syndrome type II (Schmidt syndrome). Praxis (Bern 1994). 2008 Jan 23;97(2):77-81. [↩]
Van den Driessche A, Eenkhoorn V, Van Gaal L, De Block C. Type 1 diabetes and autoimmune polyglandular syndrome: a clinical review. Neth J Med. 2009 Dec;67(11):376-87. [↩]
Microscopic View of Hemochromatosis Stained Blue. Courtesy Wikimedia
What Is Hemochromatosis?
[dropcap]H[/dropcap]emochromatosis, also called iron overload liver disease, is a common inherited disease in the Caucasian population that is characterized by increased iron deposition within the tissues (overload) associated with injury to them.
Hemochromatosis is an autosomal recessive disease, meaning a pair of abnormal genes are inherited from each parent. Parents are unaffected because they carry only one gene.
Q: Where is iron deposited?
A: In hemochromatosis, more iron is absorbed from the small intestine than is needed by the body. Subsequently, because the body has no satisfactory means to release iron overload, excess iron is deposited in various organs such as the liver causing cirrhosis, joints causing arthritis, and the pancreas causing diabetes mellitus.1
Consumption of alcoholic drinks with food should be avoided because alcohol increases leaky gut and greatly increases the absorption of iron. On the flip side, eggs and foods containing calcium such as milk and cheese are beneficial for hemochromatosis because they impair the absorption of iron. Another iron inhibitor is food that contains phytic acid which includes the bran and outer layer of grains, seeds, nuts, peas, beans, and lentils.
Iron overloading, as measured by a random (non-fasting) elevated transferrin saturation value, is estimated to occur in 1 to 6 people per 100 in the United States according to the CDC (Center for Disease Control and Prevention). Too much iron increases the risk for metabolic syndrome, type 2 diabetes mellitus, cancer, liver disease, and osteoporosis.
Medical treatment to remove excess iron from the body uses phlebotomy that draws blood through a vein and/or certain injectable drugs (deferoxamine and deferairox) that bind to iron.
What Is Hemochromatosis In Celiac Disease and/or Gluten Sensitivity?