[dropcap]A[/dropcap]nti-tissue transglutaminase antibodies (anti-tTG) are connective tissue autoantibodies and can be detected in blood samples from affected persons who are reacting to gluten in the diet.
Autoantibodies are abnormal because they attack the body’s own tissue, which in the case of these antibodies is tissue transglutaminase 2 (TG2).
Q: What is tissue transglutaminase 2 (TG2)?
A: Tissue transglutaminase 2 (TG2) is an enzyme that appears in many cell locations and is particularly abundant in endothelial cells that line the small intestine. It has been implicated in a variety of cellular processes, such as differentiation, cell death, inflammation, cell migration and wound healing.
The cell appears to adapt the dynamics of this enzyme to meet specific sub-cellular needs or to respond to stress or other stimuli. Substantial evidence indicates that the location of TG2 within cells is critical for the regulation of its various biochemical activities, which subsequently trigger diverse downstream events,1
Although initially studied as an enzyme within cells, TG2 is now known to be secreted also into the extracellular space (between cells) or onto the cell surface.1
Abnormal activation of TG2 or deregulation of its function(s) is involved in a variety of human diseases, such as celiac disease, diabetes, neurodegenerative diseases, multiple sclerosis and rheumatoid arthritis. A role in inflammatory disorders and septic shock has also been shown. Moreover, multiple studies have revealed elevated TG2 expression in many types of cancer cells.1
What Are Anti-tissue Transglutaminase Antibodies In Celiac Disease and/or Gluten Sensitivity?
Sources:
Piacentini M, D’Eletto M, Farrace MG, Rodolfo C, Del Nonno F, Ippolito G, Falasca L. Characterization of distinct sub-cellular location of transglutaminase type II: changes in intracellular distribution in physiological and pathological states. Cell Tissue Res. 2014 Dec;358(3):793-805. doi: 10.1007/s00441-014-1990-x. [↩] [↩] [↩]
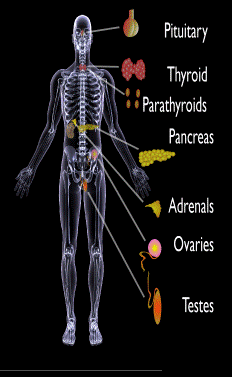
Endocrine glands targeted in polyglandular autoimmune syndrome.
What Are Autoimmune Polyglandular Syndromes?
[dropcap]A[/dropcap]utoimmune polyglandular syndromes (APS) are rare clusterings of two or more endocrine and non-endocrine autoimmune disorders in the same affected person.
Polyglandular is somewhat of a misnomer since many of the manifestations of the diseases do not concern endocrine glands.1
Endocrine autoimmune disorders involve the abnormal production of autoantibodies that target and destroy the body’s own endocrine tissues, causing loss of essential hormone production by the targeted glands. Endocrine glands include the pituitary, thyroid, adrenal, parathyroid, islets of Langerhans (pancreas), testes in males, and ovaries in females.
First degree relatives (siblings of same parents, parents, children) have an increased incidence of latent, meaning not apparent, autoimmune pathology.2
Q: How many autoimmune polyglandular syndromes are described?
A:Three syndromes have been identified and they are all inherited: APS type-1, APS type-2, and APS type-3.
APS type-1 is a genetic mutation inherited in an autosomal recessive manner. A child with APS type-1 has inherited two mutated copies of a gene called the AIRE (autoimmune regulator) gene, which is on the long arm of 21st chromosome present in each cell.3 The parents, called carriers, are unaffected since they each have only a single copy of the AIRE mutated gene. Humans have a total of 23 pairs of chromosomes that contain genes inherited from each parent. Mutations in the genes cause disease.
Diagnosis criteria for autoimmune polyglandularsyndrome type-1 includes these three disorders:
Chronic candida infection (CMC), which usually develops first, typically attacks skin, but very commonly also nails, mouth, vagina, esophagus and intestine. CMC in APS type-1 patients is usually mild, and in most cases, it is chronic. It is found in 73–100 % of APS type-1 patients.
Hypoparathyroidism, causing loss of parathyroid function (hypoparathyreosis) is found in 76–93 % of APS type-1 patients.
Autommune Addison’s disease, also called autoimmune adrenalitis, is found in 72-100 % of APS type-1 patients. Still many of them die for unrecognized or late diagnosed autoimmune Addison’s disease, so regular follow-up for children in suspicion of APS type-1 (with CMC or/and hypoparathyroidism) is necessary.4
Other assocated disorders that may develop, but are not required for diagnosis, include: vitiligo, premature menopause, pernicious anemia, parathyroid gland failure, alopecia, and celiac disease. Thyroid disease rarely occurs.5
APS type-2 is linked to the inheritance of HLA antigens on chromosome 6 and appears to be autosomal dominant with incomplete penetrance. This suggests the contribution of environmental factors, such as bacterial and viral infections, medications, psychological factors, etc.6It does not have an identified mutation of the AIRE gene.
Diagnosis criteria for autoimmune polyglandular syndrome type-2 includes these two disorders:
Autommune Addison’s disease combined with
Autoimmune thyroid disease (thyroid atrophy, hypertrophic goiter related to Hashimoto’s thyroiditis, Graves’ disease, asymptomatic autoimmune thyroiditis)6 and/or type I diabetes mellitus. The conditions may occur in any order.
Polyglandular autoimmune syndrome type-2 is also known as Schmidt’s syndrome when adrenalitis (adrenal insufficiency) is associated with thyroiditis and Carpenter’s syndrome for adrenal insufficiency with hypoparathyreosis (impaired function of parathyroid glands).
Other disorders that may develop, but are not required for diagnosis, include: type 1 diabetes (50%), frequently gonadal failure or vitiligos, also celiac disease, autoimmune hepatitis, alopecia, pernicious anemia, and myasthenia gravis.7 Decades may arise between the onset of one disease and the onset of the second in the same patient.8
Therapy of APS type-2 consists of hormone replacement therapy for each separate condition, except that treatment for adrenal insufficiency must be given before thyroid therapy is started when the conditions occur together.9 Thyroxin replacement may induce life-threatening adrenal failure in a patient with untreated Addison’s disease. Thus, in case of doubt hydrocortisone should be given before the thyroxine administration is started.10
APS type-3 has a strong genetic background. Diagnosis criteria for autoimmune polyglandular syndrome type-3 involves these conditions:
Autoimmune thyroiditis that occurs with another organ-specific autoimmune disease, but not with autoimmune Addison’s disease, and
Other autoimmune diseases can include diabetes mellitus, pernicious anemia, vitiligo, alopecia, myasthenia gravis, celiac disease, and Sjögren’s syndrome. The most common APS type-3 combination is autoimmune disease of thyroid gland and pernicious anemia.11
Who is Affected in the General Population?
APS type-1 is usually apparent in childhood with the incidence of 1 in100,000 persons. It is more common among Finns (1 in 25,000), Sardinians (1 in 14,000), and Iranian Jews (1 in 6,500 to 1 in 9,000). The age of onset is usually early childhood, but new symptoms can develop throughout life. It affects both sexes equally.
APS type-2 has a peak onset in middle age, although the first signs usually develop between 20–30 years of age. Its prevalence is 1 in 20,000 persons. It is three times more frequent among women than men.12
APS type-3 is most frequent among middle-aged women.7[/box]
What Is Autoimmune Polyglandular Syndrome In Celiac Disease and/or Gluten Sensitivity?
Sources:
Wémeau JL, Proust-Lemoine E, Ryndak A, Vanhove L. Thyroid autoimmunity and polyglandular endocrine syndromes. Hormones (Athens). 2013 Jan-Mar;12(1):39-45. [↩]
Femiano P, Castaldo V, Iossa C. Complex family association in autoimmune polyendocrine syndrome. Minerva Pediatrica. Apr 2003;55(2):163-70. [↩]
Wémeau JL, Proust-Lemoine E, Ryndak A, Vanhove L. Thyroid autoimmunity and polyglandular endocrine syndromes. Hormones (Athens). 2013 Jan-Mar;12(1):39-45. [↩]
Wémeau JL, Proust-Lemoine E, Ryndak A, Vanhove L. Thyroid autoimmunity and polyglandular endocrine syndromes. Hormones (Athens). 2013 Jan-Mar;12(1):39-45. [↩]
Wémeau JL, Proust-Lemoine E, Ryndak A, Vanhove L. Thyroid autoimmunity and polyglandular endocrine syndromes. Hormones (Athens). 2013 Jan-Mar;12(1):39-45. [↩] [↩]
MAJERONI BA and PATEL P. Autoimmune Polyglandular Syndrome, Type II. Am Fam Physician. 2007 Mar 1;75(5):667-670. [↩]
Lipowsky C, Schorl-Schweikardt BA, Kehl O, Brändle M. 19-year-old patient with adrenal cortex insufficiency–only the tip of the iceberg. Polyendocrine autoimmune syndrome type II (Schmidt syndrome). Praxis (Bern 1994). 2008 Jan 23;97(2):77-81. [↩]
Van den Driessche A, Eenkhoorn V, Van Gaal L, De Block C. Type 1 diabetes and autoimmune polyglandular syndrome: a clinical review. Neth J Med. 2009 Dec;67(11):376-87. [↩]
[dropcap]C[/dropcap]hronic bullous dermatosis of childhood, also termed linear IgA dermatosis, is the most common acquired autoimmune blistering disorder of childhood and is characterized by itchy, urticated papules and plaques as well as polycyclic lesions (merged circles) with blisters at the edge, located on normal looking skin around the mouth and perineum in young children. In children over 7 years, other parts of the body may rather be affected.
Q: What tissue is targeted in chronic bullous dermatosis of childhood?
A: In chronic bullous dermatosis of childhood, there is an autoimmune attack on structural proteins, usually proteolytic fragments of collagen XVII, which renders the dermal-epidermal junction prone to blistering.
The dermal-epidermal junction is where the surface skin layer, or epidermis, meets the lower layer, or dermis. Diagnosis is confirmed by characteristic histology and direct immunofluorescence showing linear IgA (immunoglobulin A antibody) staining of the basement membrane zone.1
The incidence of chronic bullous disease of childhood is rare. Age of onset is typically before 5 years of age and is seen in all ethnic groups.
What Is Chronic Bullous Dermatosis Of Childhood In Celiac Disease and/or Gluten Sensitivity?
Sources:
Mintz EM, Morel KD.Clinical features, diagnosis, and pathogenesis of chronic bullous disease of childhood. Dermatol Clin. 2011 Jul;29(3):459-62, ix. doi: 10.1016/j.det.2011.03.022. [↩]
Microscopic view of pancreatic islet cells. Courtesy Dr. José Sánchez Gonzales
What Is Type I Diabetes Mellitus?
[dropcap]T[/dropcap]ype 1 diabetes mellitus (T1DM), also termed type 1A, is an inherited autoimmune disorder in which anti-islet autoantibodies destroy the islet cells of the pancreas that secrete insulin hormone. Type 1 diabetes mellitus was formerly called juvenile diabetes because it usually afflicts persons under the age of 25 years.
Loss of insulin production results in failure to metabolize glucose. Glucose is a simple sugar that is a required source of energy for the body, especially the brain and muscles.
Type 1 diabetes mellitus is characterized by sustained fasting blood glucose levels above 126 mg/dL (hyperglycemia) with subsequent loss of glucose from the body by removal through the urine (glucosuria) as the body attempts to lower blood glucose, and cell starvation that follows.
That is, while glucose accumulates in blood, the body cannot access it. Without insulin treatment, this disorder quickly produces coma and ultimately results in death. In fact, it is 5th leading cause of death in the United States.
Q: How does insulin work?
A: Insulin moves glucose from the bloodstream into body cells where it is used or reformulated for high energy storage. For example, muscles can use glucose for immediate work or store it in the form of glygogen for later work, depending on need. Healthy insulin production keeps an 8 hour fasting blood glucose level to less than 100 mg/dL. Upon eating carbohydrate food, glucose is digested and absorbed from the small intestine into the bloodstream which then raises blood glucose levels. The elevated level is controlled by prompt action of insulin to lower it to below 140 mg/dL within 2 hours of eating.
Insulin does not work alone. The islets of Langerhans manage glucose in the body. The islets are specialized formations located on the outer surface of the pancreas. The islets are composed of two different types of cells known as alpha and beta cells. These cells make the competing hormones that keep blood glucose within a healthy range.
Alpha cells secrete glucagon to raise blood glucose levels by triggering the body to release stored energy in the form of glycogen. In the opposite, beta cells secrete insulin to lower blood glucose by opening body cells so that glucose in blood can enter. Without insulin, glucose cannot enter cells but remains in the bloodstream where it accumulates.
Insulin is also needed to move magnesium into cells from the bloodstream. On the other side, magnesium is needed to produce insulin. Insulin has other functions such as building muscle and helping regulate cholesterol which directly impacts the sex hormones, estrogen, progesterone, and testosterone.
Onset of symptoms usually occurs over a period of days or weeks, although beta cell destruction can begin years earlier. The SEARCH for Diabetes in Youth multicenter study, funded by the Centers for Disease Control and Prevention (CDC) and the National Institutes of Health (NIH), has determined that based on data from 2002 to 2003, a total of 15,000 youth in the United States were newly diagnosed with type 1 diabetes each year. Non-Hispanic white youth had the highest rate of new cases of type 1 diabetes according to NIH.
Type 1A diabetes mellitus has become one of the most intensively studied autoimmune disorders. It is now possible to predict its development, beginning with HLA-encoded genetic susceptibility, followed by the development of a series of anti-islet autoantibodies.1
What Is Type I Diabetes Mellitus In Celiac Disease and/or Gluten Sensitivity?
Sources:
Liu E, Eisenbarth GS. Type 1A diabetes mellitus-associated autoimmunity. Endocrinology and Metabolism Clinics of North America. Jun 2002;31(2):391-410, vii-viii. [↩]
An autoimmune, chronic slowly progressive demyelinating (damage to nerve fibers) disease of the central nervous system characterized by multiple and varied neurologic symptoms and signs such as numbness, muscle weakness and visual disturbances, usually with…
An autoimmune disorder causing chronic dryness with inflammation of the conjunctiva and cornea of both eyes due to tear abnormality. Click for full description.
Cancel your subscription?
We're sorry to see you go.
Please select a reason for cancellation.
Please provide additional details.
Cancellation request submitted
We've received your request. Your subscription remains active while we process it. We'll email you once it's confirmed.
Cancellation pending
Access Content
Affiliate Terms
By joining our Affiliate Program, you agree to the following terms:
1. Eligibility: You must be 18 years or older to participate. We reserve the right to refuse service to anyone for any reason at any time.
2. Promotional Guidelines: Affiliates must not engage in misleading advertising or spam. Promotions must accurately represent our brand and products.
3. Affiliate Links: You must use your unique tracking link to receive credit for referred sales.
4. Prohibited Use: You may not use our branding in a way that may confuse customers or misrepresent your relationship with us.
5. Termination: We reserve the right to terminate your account at any time for breach of these terms.
6. Changes to Agreement: We may update these terms at any time. Continued participation implies acceptance of any changes.
By continuing, you agree to the terms of this Affiliate Agreement.