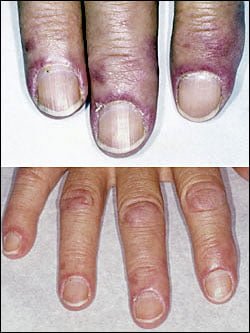
[dropcap]I[/dropcap]ron deficiency anemia is a blood cell disorder that is characterized by formation of small, pale red blood cells, causing tissue hypoxia. Hypoxia is the inability to meet the demands of the body for oxygen.
Q: Why do small, pale red blood cells cause tissue hypoxia?
A: Small, pale red blood cells (erythrocytes) cause tissue hypoxia because they are not able, as do normal erythrocytes, to pick up adequate oxygen from the lungs and carry it to cells that use oxygen.
Red blood cell production and function are dependent on a sufficient level of iron in the body and also the ability to use available iron to make hemoglobin in red blood cells.
Hemoglobin is a protein that binds oxygen in red blood cells to be carried by the bloodstream to cells throughout the body. In iron deficiency anemia, hemoglobin in females is below 12.5g/dl (normal range is 12.5 to 16g/dl) and in males it is below 13.5g/dl (normal range is 13.5 to 17.5g/dl).
Iron must be obtained from the diet, since the body cannot make it, but there are various factors that can interfere with absorption and use in the body, causing anemia. Iron absorption from the gut first requires ionization, or gaining a positive electrical charge, in the strongly acidic environment of stomach juice. Ionized iron, only, can be absorbed in the duodenum, which receives the acidic contents of the stomach before it is neutralized further along.
Dietary iron can be heme or non-heme depending on the food source. Heme iron obtained only from animal food sources is absorbed into the bloodstream by active transport across the brush border (microvilli) which cover the multitudinous villi of the small intestinal lining.
Non-heme iron obtained from plants must bind with apoprotein after entering the enterocyte (surface cell of small intestinal lining) to be ferried to the underlying basolateral membrane and exited by active transport into the bloodstream.
Frequently, chronic anemia due to iron deficiency is accompanied by increased platelets, and this thrombocytosis resolves with iron repletion (normal iron level). Conversely, in severe iron deficiency anemia, patients may have thrombocytopenia (low platelets), which also resolves with iron therapy.1
What Is Iron Deficiency Anemia In Celiac Disease and/or Gluten Sensitivity?
Sources:
Koury M and Rhodes M. How to approach chronic anemia. Hematology Am Soc Hematol Educ Program. 2012;2012:183-90. doi: 10.1182/asheducation-2012.1.183. [↩]
[dropcap]D[/dropcap]ermatomyositis is a rare autoimmune systemic disease of the connective tissue that is characterized by inflammatory and debilitatingdegenerative changes in the muscles and in the skin.
Dermatomyositis results in symmetric, proximal muscle weakness of limbs (upper arms and legs), and skin manifestations. 50-70% of patients have circulating myositis-specific auto-antibodies.
The course of dermatomyositis is unpredictable being marked by spontaneous flare-ups and remissions. It can begin slowly or abruptly according to the factor that is triggering the onset such as infection, medications like phenytoin, and autoimmune disease.
Q: What are the skin manifestations of dermatomyositis?
A: Classic skin manifestations of dermatomyositis include these features:
The heliotrope rash (lilac color) on upper eyelids.
Rash on face, neck, shoulders, upper chest, elbows, knees, knuckles, and back.
Gottron’s papules (scaly, red eruptions or patches over the knuckles, elbows, and knees).
The V-sign (rash front of neck and chest).
The shawl sign (rash distribution on shoulders and back).1
Additional cutaneous manifestations are described below under symptoms.
Dermatomyositis is associated with an increased risk of cancer, other autoimmune diseases, such as lupus and psoriasis, and it can be a complication of interferon-α therapy.About 1 person in 100,000 are affected according to various studies. While it affects all ages, women have twice the occurence of men.
There is no cure for dermatomyositis, but the symptoms can be treated. Options include medication, physical therapy, exercise, heat therapy (including microwave and ultrasound), orthotics and assistive devices, and rest. The standard treatment for dermatomyositis is a corticosteroid drug, given either in pill form or intravenously. Immunosuppressant drugs, such as azathioprine and methotrexate, may reduce inflammation in people who do not respond well to prednisone.2
What Is Dermatomyositis In Celiac Disease and/or Gluten Sensitivity?
Sources:
Marvi U, Chung L, Fiorentino DF. Clinical presentation and evaluation of dermatomyositis. Indian J Dermatol. 2012 Sep;57(5):375-81. doi: 10.4103/0019-5154.100486. [↩]
National Institute of Neurological Disorders and Stroke. [↩]
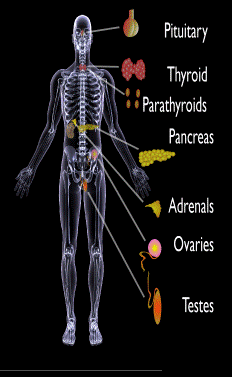
Endocrine glands targeted in polyglandular autoimmune syndrome.
What Are Autoimmune Polyglandular Syndromes?
[dropcap]A[/dropcap]utoimmune polyglandular syndromes (APS) are rare clusterings of two or more endocrine and non-endocrine autoimmune disorders in the same affected person.
Polyglandular is somewhat of a misnomer since many of the manifestations of the diseases do not concern endocrine glands.1
Endocrine autoimmune disorders involve the abnormal production of autoantibodies that target and destroy the body’s own endocrine tissues, causing loss of essential hormone production by the targeted glands. Endocrine glands include the pituitary, thyroid, adrenal, parathyroid, islets of Langerhans (pancreas), testes in males, and ovaries in females.
First degree relatives (siblings of same parents, parents, children) have an increased incidence of latent, meaning not apparent, autoimmune pathology.2
Q: How many autoimmune polyglandular syndromes are described?
A:Three syndromes have been identified and they are all inherited: APS type-1, APS type-2, and APS type-3.
APS type-1 is a genetic mutation inherited in an autosomal recessive manner. A child with APS type-1 has inherited two mutated copies of a gene called the AIRE (autoimmune regulator) gene, which is on the long arm of 21st chromosome present in each cell.3 The parents, called carriers, are unaffected since they each have only a single copy of the AIRE mutated gene. Humans have a total of 23 pairs of chromosomes that contain genes inherited from each parent. Mutations in the genes cause disease.
Diagnosis criteria for autoimmune polyglandularsyndrome type-1 includes these three disorders:
Chronic candida infection (CMC), which usually develops first, typically attacks skin, but very commonly also nails, mouth, vagina, esophagus and intestine. CMC in APS type-1 patients is usually mild, and in most cases, it is chronic. It is found in 73–100 % of APS type-1 patients.
Hypoparathyroidism, causing loss of parathyroid function (hypoparathyreosis) is found in 76–93 % of APS type-1 patients.
Autommune Addison’s disease, also called autoimmune adrenalitis, is found in 72-100 % of APS type-1 patients. Still many of them die for unrecognized or late diagnosed autoimmune Addison’s disease, so regular follow-up for children in suspicion of APS type-1 (with CMC or/and hypoparathyroidism) is necessary.4
Other assocated disorders that may develop, but are not required for diagnosis, include: vitiligo, premature menopause, pernicious anemia, parathyroid gland failure, alopecia, and celiac disease. Thyroid disease rarely occurs.5
APS type-2 is linked to the inheritance of HLA antigens on chromosome 6 and appears to be autosomal dominant with incomplete penetrance. This suggests the contribution of environmental factors, such as bacterial and viral infections, medications, psychological factors, etc.6It does not have an identified mutation of the AIRE gene.
Diagnosis criteria for autoimmune polyglandular syndrome type-2 includes these two disorders:
Autommune Addison’s disease combined with
Autoimmune thyroid disease (thyroid atrophy, hypertrophic goiter related to Hashimoto’s thyroiditis, Graves’ disease, asymptomatic autoimmune thyroiditis)6 and/or type I diabetes mellitus. The conditions may occur in any order.
Polyglandular autoimmune syndrome type-2 is also known as Schmidt’s syndrome when adrenalitis (adrenal insufficiency) is associated with thyroiditis and Carpenter’s syndrome for adrenal insufficiency with hypoparathyreosis (impaired function of parathyroid glands).
Other disorders that may develop, but are not required for diagnosis, include: type 1 diabetes (50%), frequently gonadal failure or vitiligos, also celiac disease, autoimmune hepatitis, alopecia, pernicious anemia, and myasthenia gravis.7 Decades may arise between the onset of one disease and the onset of the second in the same patient.8
Therapy of APS type-2 consists of hormone replacement therapy for each separate condition, except that treatment for adrenal insufficiency must be given before thyroid therapy is started when the conditions occur together.9 Thyroxin replacement may induce life-threatening adrenal failure in a patient with untreated Addison’s disease. Thus, in case of doubt hydrocortisone should be given before the thyroxine administration is started.10
APS type-3 has a strong genetic background. Diagnosis criteria for autoimmune polyglandular syndrome type-3 involves these conditions:
Autoimmune thyroiditis that occurs with another organ-specific autoimmune disease, but not with autoimmune Addison’s disease, and
Other autoimmune diseases can include diabetes mellitus, pernicious anemia, vitiligo, alopecia, myasthenia gravis, celiac disease, and Sjögren’s syndrome. The most common APS type-3 combination is autoimmune disease of thyroid gland and pernicious anemia.11
Who is Affected in the General Population?
APS type-1 is usually apparent in childhood with the incidence of 1 in100,000 persons. It is more common among Finns (1 in 25,000), Sardinians (1 in 14,000), and Iranian Jews (1 in 6,500 to 1 in 9,000). The age of onset is usually early childhood, but new symptoms can develop throughout life. It affects both sexes equally.
APS type-2 has a peak onset in middle age, although the first signs usually develop between 20–30 years of age. Its prevalence is 1 in 20,000 persons. It is three times more frequent among women than men.12
APS type-3 is most frequent among middle-aged women.7[/box]
What Is Autoimmune Polyglandular Syndrome In Celiac Disease and/or Gluten Sensitivity?
Sources:
Wémeau JL, Proust-Lemoine E, Ryndak A, Vanhove L. Thyroid autoimmunity and polyglandular endocrine syndromes. Hormones (Athens). 2013 Jan-Mar;12(1):39-45. [↩]
Femiano P, Castaldo V, Iossa C. Complex family association in autoimmune polyendocrine syndrome. Minerva Pediatrica. Apr 2003;55(2):163-70. [↩]
Wémeau JL, Proust-Lemoine E, Ryndak A, Vanhove L. Thyroid autoimmunity and polyglandular endocrine syndromes. Hormones (Athens). 2013 Jan-Mar;12(1):39-45. [↩]
Wémeau JL, Proust-Lemoine E, Ryndak A, Vanhove L. Thyroid autoimmunity and polyglandular endocrine syndromes. Hormones (Athens). 2013 Jan-Mar;12(1):39-45. [↩]
Wémeau JL, Proust-Lemoine E, Ryndak A, Vanhove L. Thyroid autoimmunity and polyglandular endocrine syndromes. Hormones (Athens). 2013 Jan-Mar;12(1):39-45. [↩] [↩]
MAJERONI BA and PATEL P. Autoimmune Polyglandular Syndrome, Type II. Am Fam Physician. 2007 Mar 1;75(5):667-670. [↩]
Lipowsky C, Schorl-Schweikardt BA, Kehl O, Brändle M. 19-year-old patient with adrenal cortex insufficiency–only the tip of the iceberg. Polyendocrine autoimmune syndrome type II (Schmidt syndrome). Praxis (Bern 1994). 2008 Jan 23;97(2):77-81. [↩]
Van den Driessche A, Eenkhoorn V, Van Gaal L, De Block C. Type 1 diabetes and autoimmune polyglandular syndrome: a clinical review. Neth J Med. 2009 Dec;67(11):376-87. [↩]
Cancel your subscription?
We're sorry to see you go.
Please select a reason for cancellation.
Please provide additional details.
Cancellation request submitted
We've received your request. Your subscription remains active while we process it. We'll email you once it's confirmed.
Cancellation pending
Access Content
Affiliate Terms
By joining our Affiliate Program, you agree to the following terms:
1. Eligibility: You must be 18 years or older to participate. We reserve the right to refuse service to anyone for any reason at any time.
2. Promotional Guidelines: Affiliates must not engage in misleading advertising or spam. Promotions must accurately represent our brand and products.
3. Affiliate Links: You must use your unique tracking link to receive credit for referred sales.
4. Prohibited Use: You may not use our branding in a way that may confuse customers or misrepresent your relationship with us.
5. Termination: We reserve the right to terminate your account at any time for breach of these terms.
6. Changes to Agreement: We may update these terms at any time. Continued participation implies acceptance of any changes.
By continuing, you agree to the terms of this Affiliate Agreement.